【临床研究】浅析药物临床试验中AE/SAE/SUSAR的管理要点
来源:临床医学研究中心 发布时间:2022年7月20日
药物临床试验是指以人体(患者或健康受试者)为对象的试验,意在发现或验证某种试验药物的临床医学、药理学以及其他药效学作用、药物不良反应或者试验药物的吸收、分布、代谢和排泄,以确定药物的疗效与安全性的系统性试验。
临床试验阶段应明确对试验药物安全性事件的定义、收集和分析处理,除满足法规要求上报严重不良事件及可疑非预期严重不良反应 (SUSAR),申办者还应依据该试验药物的安全性特征,在试验方案中定义并收集该药物需特殊关注的不良事件。在临床试验过程中,除需关注有效性评价外,同时亦需注重安全性评价,该评价是起草和撰写药品说明书安全性信息(如药品不良反应、禁忌、注意事项等)的重要依据,亦可为上市后临床合理用药提供重要的安全保障。
1、定义
不良事件(adverse event,AE),指受试者接受试验用药品后出现的所有不良医学事件,可以表现为症状体征、疾病或者实验室检查异常,但不一定与试验用药品有因果关系。
严重不良事件(SAE),指受试者接受试验用药品后出现死亡、危及生命、永久或者严重的残疾或者功能丧失、受试者需要住院治疗或者延长住院时间,以及先天性异常或者出生缺陷等不良医学事件。
可疑且非预期严重不良反应(SUSAR),指临床表现的性质和严重程度超出了试验药物研究者手册、已上市药品的说明书或者产品特性摘要等已有资料信息的可疑并且非预期的严重不良反应。
2、不良事件分级
一般采用以下2种:
1.根据AE的严重程度,将AE划分为轻、中、重3级。轻度:容易耐受,不需要治疗,且不影响受试者日常活动的事件。中度:导致轻微不便或需要给予治疗措施,且影响受试者日常活动的事件。重度:需要全身药物治疗或其他治疗,对受试者日常活动有重大影响,且可能致残的事件。
2.参考美国国立癌症研究所常见不良反应事件评价标准(National Cancer Institute Common Terminology Criteria for Adverse Events,NCI CTCAE)、世界卫生组织(World Health Organization,WHO)等分级标准,根据AE的严重程度进行5级划分。
1级:轻度,无临床症状或有轻微临床症状,或仅有临床或实验室检查异常,无需进行干预,不需对症处理,不需停药。
2级:中度,需要最小的、局部的或无创伤的干预,或日常生活活动受限,主诉不适,需对症处理,不需停药。
3级:严重或者具重要医学意义但暂时不会危及生命,导致住院或延长住院时间,致残,日常生活自理受限,主诉明显不适,需对症处理,并需暂停用药。
4级:危及生命,需要紧急干预。
5级:与AE相关的死亡。
3、AE/SAE因果关系判断标准
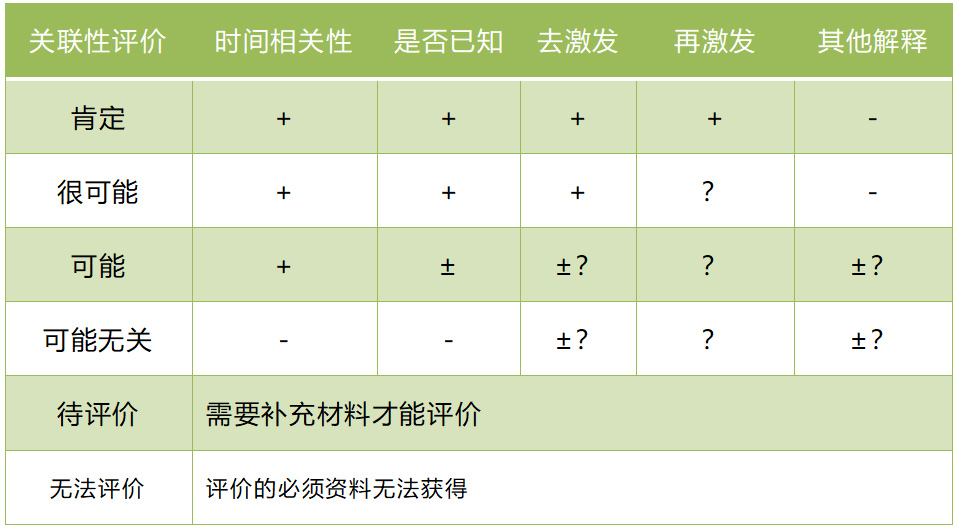
因果关系判断有多种可用的方法,比如:Karch 和 Lasagna 法,Naranjo 法,但并没有一个金标准。根据我国《个例药品不良反应收集和报告指导原则》(2018 年第 131 号),关联性评价分为肯定、很可能、可能、可能无关、待评价、无法评价 6 级,但该指导原则是为规范药品上市后个例不良反应的收集和报告而颁布,新药临床试验中采用可能存在一定局限性,仅供申办者和研究者参考,标准见表 1。
▲表 1 不良事件/严重不良事件的因果关系判断标准
注:“+”表示肯定或阳性;“-”表示否定或阴性;“±”表示难以判断;“?”表示不明。时间相关性:用药与不良反应的出现有无合理的时间关系。是否已知:不良反应是否符合该药已知的不良反应类型。去激发:停药或减量后,不良反应是否消失或减轻。再激发:再次使用可疑药品是否再次出现同样的不良反应。其他解释:不良反应是否可用并用药品的作用、患者病情的进展、其他治疗的影响来解释。
如果对照表 1 中的 5 条标准,不能完全对应某条可能性时,建议采用保守原则或称之为不利于新药原则,即如果判断结果介于“很可能相关”与“可能相关”之间,应该判“很可能相关”或在信息不足的情况下评估为“可能相关”。
当相关性选项中有“无法评价/判断”时,仅在因客观原因无法获得进一步信息,不足以判断因果关系时,才可选择这一选项。
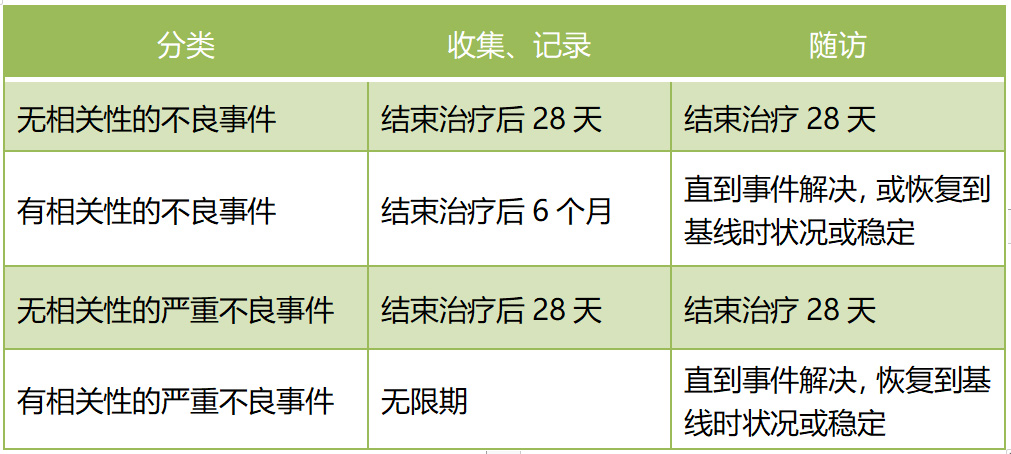
4、严重不良事件处理原则、报告及随访时限
(一)处理原则
1. 首先应保证受试者得到及时、适当的临床诊治。
2. 其次积极收集相关资料,如诊疗记录、检查检验结果,以便准确和及时填写严重不良事件报告,并向申办者报告。
3.确保报告与原始记录、CRF以及其他试验记录一致。确保严重不良事件的起止日期和主要的事件描述与CRF和其他试验文件一致。确保报告与原始资料、CRF中合并用药的记录,如药品名称和使用(起止日期、剂量、途径、频次)的描述,也应是一致的。
4.申办者收到任何来源的安全性相关信息后,均应当立即分析评估,包括严重性、与试验药物的相关性以及是否为预期事件等。申办者在评估事件的严重性和相关性时,如与研究者持不同意见,特别是对研究者的判断有降级的意见,须写明理由。在相关性判断中不能达成一致时,其中任一方判断不能排除与试验药物有关的,也应快速报告。
(二)报告时限
1. 报告时限从研究者获知事件发生时开始计时,填写严重不良事件表格时一般需填写获知时间。
2. 研究者按照研究方案或标准操作规程(SOP)中规定的报告方式,立即将严重不良事件(一般为获知后24小时内)报告给申办者。
(三)随访时限
注:药物临床试验安全性评价是临床试验过程中重要一环,研究者在实践中可同时参考相关法规及院内管理制度执行,尽量避免收集遗漏、漏报的情况发生。
参考资料:
1. 国家药品监督管理局,国家卫生健康委员会.关于发布药物临床试验质量管理规范的公告(2020年第57号).
2.药监局2018年4月27日发布的《药物临床试验期间安全性数据快速报告标准和程序》
3.药物临床试验安全评价.广东共识(2020年版)

 020-87001380
020-87001380


